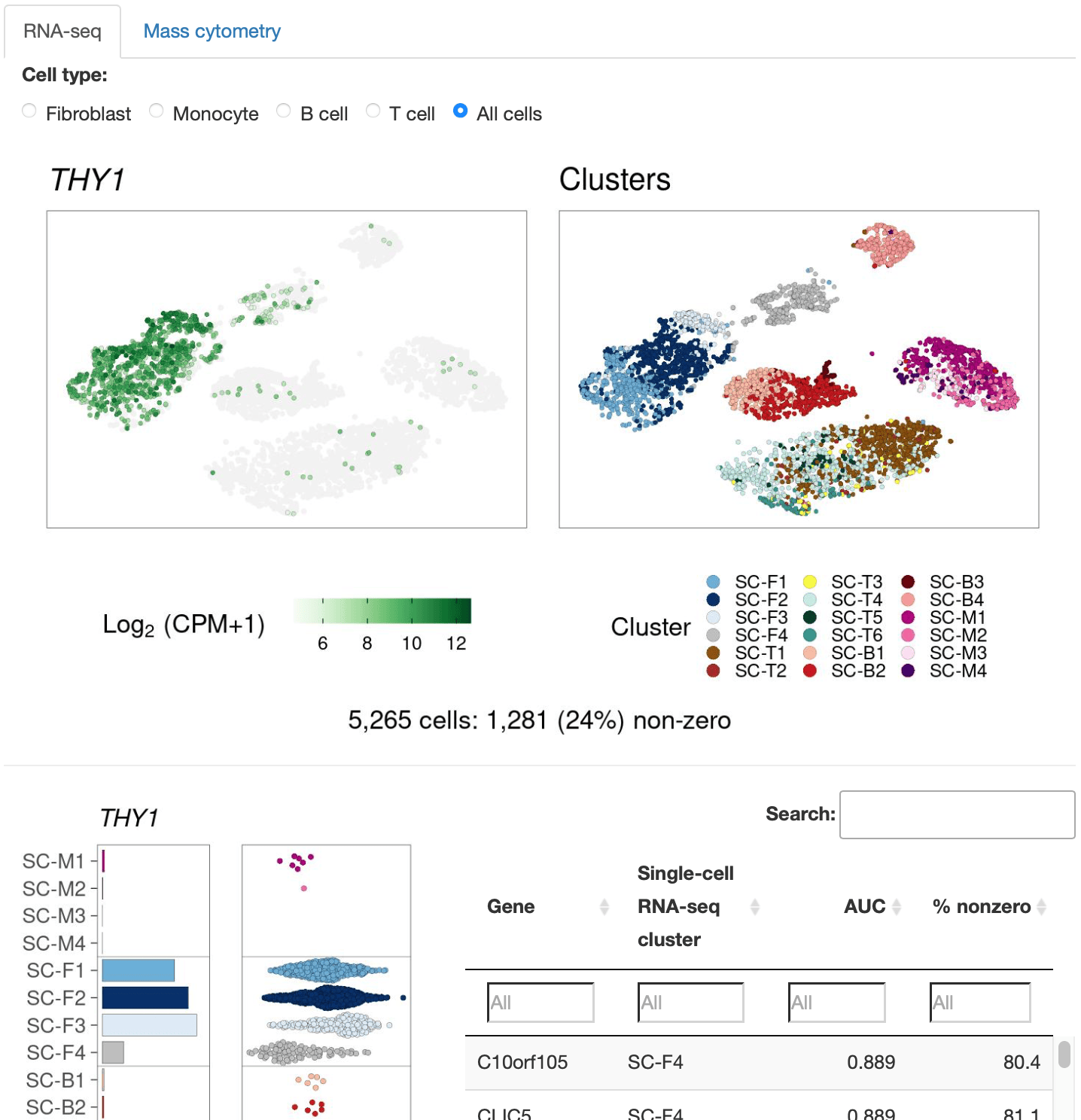
Cell Clusters
Metadata variables and gene expression in two-dimensional embeddings.
Launch Viewer →Defining Inflammatory Cell States in Rheumatoid Arthritis Joint Synovial Tissues by Integrating Single-cell Transcriptomics and Mass Cytometry
Nature Immunology (2019) · doi:10.1038/s41590-019-0378-1
Abstract
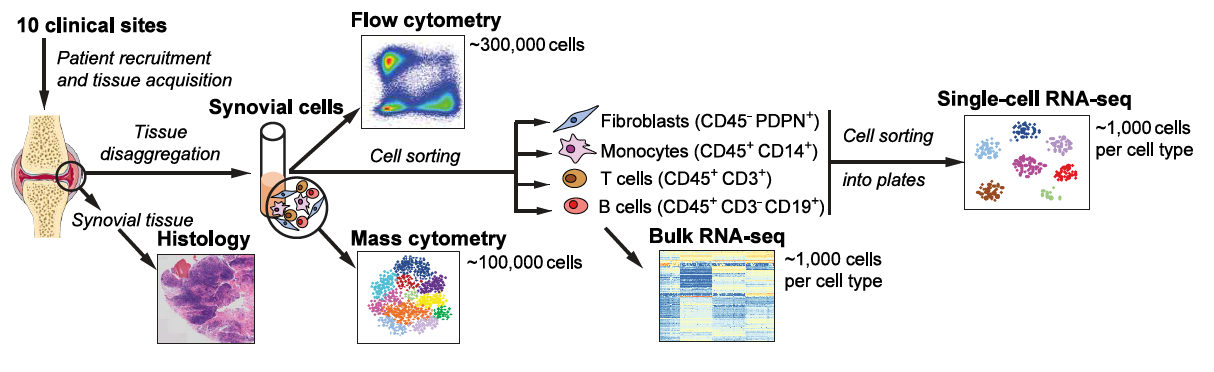
To define the cell populations that drive joint inflammation in rheumatoid arthritis (RA), we applied single-cell RNA sequencing (scRNA-seq), mass cytometry, bulk RNA sequencing (RNA-seq) and flow cytometry to T cells, B cells, monocytes, and fibroblasts from 51 samples of synovial tissue from patients with RA or osteoarthritis (OA). Utilizing an integrated strategy based on canonical correlation analysis of 5,265 scRNA-seq profiles, we identified 18 unique cell populations. Combining mass cytometry and transcriptomics revealed cell states expanded in RA synovia: THY1(CD90)+HLA-DRAhi sublining fibroblasts, IL1B+ pro-inflammatory monocytes, ITGAX+TBX21+ autoimmune-associated B cells and PDCD1+ peripheral helper T (TPH) cells and follicular helper T (TFH) cells. We defined distinct subsets of CD8+ T cells characterized by GZMK+, GZMB+, and GNLY+ phenotypes. We mapped inflammatory mediators to their source cell populations; for example, we attributed IL6 expression to THY1+HLA-DRAhi fibroblasts and IL1B production to pro-inflammatory monocytes. These populations are potentially key mediators of RA pathogenesis.
Resources
Contact
Questions or comments? Get in touch.